dnaman(序列分析软件)
v9.0大小:14.7 MB 更新:2023/03/17
类别:健康医药系统:WinXP, Win7, Win8, Win10, WinAll
分类分类
大小:14.7 MB 更新:2023/03/17
类别:健康医药系统:WinXP, Win7, Win8, Win10, WinAll
dnaman是一款功能强大的序列分析软件,软件提供了序列处理和转换、顺序组装、序列搜索、序列比较和对齐、多序列比对、限制分析、蛋白质序列与翻译分析、Oligo数据库、引物分析、DNA和蛋白质数据库等多种强大的功能,而且其具备了一个完全所见所得操作界面,同时还集成了丰富的序列分析工具,能够协助生物学家以及相关研究人员更加轻松实现序列比对、绘制质粒以及设计PCR引物,这里为您分享了dnaman 9本,能够让用户免费进行使用,有需要的朋友赶紧下载吧!
DNAMAN 是一种用于分子生物学应用的一对一软件包。该软件包提供了一个集成系统,具有多功能功能,可实现高效序列分析。它还可以进行多序列比对,设计PCR引物,蛋白质序列分析或绘制质粒。
DNAMAN的速度,多功能性,准确性和高质量的表现使其成为每个分子生物学家可以依赖的基本工具之一。
序列比较和对齐
使用DNAMAN,您可以比较两种或更多种DNA或蛋白质序列。 点阵比较提供了两个序列相似性的图形视图。 序列比对允许您在两个或更多序列中找到同源区域。 您可以使用多序列比对和系统发育树分析来分析家族成员之间的同源关系。
点阵比较
点阵比较功能在点阵图中比较两个DNA序列或两个蛋白质序列。 横坐标表示一个序列,纵坐标表示另一个序列。
两个序列比对
当您将两个或多个DNA或蛋白质序列加载到序列通道中时,序列| 对齐| 激活两个序列对齐菜单。 您可以使用此功能准确地比较两个序列。 使用此功能的另一个优点是,您可以通过定义分析区域快速对齐两个DNA或蛋白质序列的任何区域。
多序列比对
DNAMAN提供快速和最佳的比对方法,用于比对大量的DNA和蛋白质序列。 多重比对功能与许多其他序列分析程序兼容。 多序列编辑器(MASED)用于处理对齐结果。 编辑器可以在图形窗口中生成同源性和系统发育树,并以不同的序列格式导出对齐结果。
树分析
MASED可以通过多重对齐生成树木。 DNAMAN计算同源矩阵并建立所有序列对之间的相关距离。 按“输出”按钮时,会出现一个弹出菜单。 您可以选择比对的距离矩阵,系统发育树或同源树。
距离矩阵。 该矩阵显示了比对中所有序列对之间的相关距离。 低值表示两个序列之间的低分歧(高同源性)。 最大距离为1.0,表明两个序列之间没有同源性。
DNA克隆
在克隆过程中,有两个主要步骤:生成Insert和Vector片段并连接它们以创建新克隆。 DNAMAN模拟该过程以产生新的克隆序列。此功能可以帮助您在实验室工作之前设计克隆策略。
重建限制图
如果您的DNA片段没有序列信息,您可能希望在进一步工作之前找到其限制图。该过程可能涉及用单酶和双酶消化DNA,使用电泳估计所有限制性片段的大小,并根据片段大小推导出限制性图谱。重建限制图可能会很复杂。当通过酶消化产生更多片段时难以增加。 DNAMAN使用动态过程来查找与消化模式匹配的限制性片段的所有可能组合。
在实验中,您应该使用两种限制酶来消化目标DNA。必须通过电泳尽可能准确地估计单次和双次消化中所有片段的大小。选择限制| Map Reconstruction命令用来打开一个对话框。将实验数据填入表格。酶A,酶B和酶A + B的总片段大小的总和必须相等。
应准确估计DNA片段的大小。您可以使用错误栏进行估算。默认错误设置为10%。如果您对估算有信心,可以减少它。
推导出的限制图显示在图形窗口中。如果从提供的数据中找到两个以上的地图,则DNAMAN仅绘制前两个地图。可能的地图数量显示在图形窗口的底部。在这种情况下,可以通过降低错误率来找到更准确的映射。
如果您有两种以上的消化酶,请分别进行每种酶组合的分析。每次分析仅使用两种酶,然后从各个地图的组合构建最终地图。
蛋白质序列与翻译分析
DNAMAN使用遗传密码表进行翻译分析。 默认情况下,使用通用代码。 DNAMAN提供了7个遗传密码表。 此外,您可以为您的工作创建特定的遗传密码。 可以选择列表中的任何代码作为默认遗传密码,其用于序列翻译的所有功能分析中。
数据信息和设置
DNAMAN中使用的数据信息,例如限制酶数据库和氨基酸属性,可由用户访问和修改。 当您成为DNAMAN的专家时,您可以进行一些修改以利用该软件。
序列编辑和转换
支持GenBank,FASTA,GFF等文件
使用命令行和脚本进行序列转换
多序列比对
支持ClustalW,GCG,GDE和其他文件
使用命令行和脚本进行多重对齐
顺序组装
具有图形界面的快速对齐算法
使用命令行和脚本进行顺序组装
PCR Primer Design
通过图形界面优化选择
批处理和脚本
序列数据库
Sqlite3数据库引擎
支持MySQL和Microsoft SQL服务器
DNA限制性分析
具有可定制酶数据库的直观用户界面
使用命令行和脚本进行限制分析
成对序列分析和点图
DNA / RNA二级结构分析
序列搜索和BLAST分析
系统发育分析
蛋白质序列分析
蛋白质二级结构预测
翻译/反向翻译/密码子优化
寡核苷酸序列分析
序列注释和转移
批处理和常规脚本
自定义菜单命令
1、下载并解压软件,双击安装程序进入如下的DNAMAN9安装许可1协议,勾选【I accept the agreement】的选项,再单击【next】进入下一步的安装。

2、选择安装文件夹,默认的安装位置为C:\Program Files (x86)\DNAMAN,支持自定义,点击【next】。
3、准备安装,点击【install】按钮即可执行安装操作。

4、弹出如下的DNAMAN安装成功的提示,取消“launch DNAMAN”以及“launch LBDraw”的勾选,点击【finish】结束安装。

5、先不要运行软件,打开“crack”文件夹,将补丁文件夹“DNAMAN.exe”复制到软件的安装目录,默认安装目录为C:\Program Files (x86)\DNAMAN。

6、弹出如下的包含同名文件提示,点击【替换目标中的文件】。

7、运行DNAMAN即可开始进行使用。
装配DNA片段
选择序列| Sequence Assembly命令显示Sequence Assembly对话框。为了获得最佳结果,应选择适当的参数来处理序列组装。装配序列的数量目前限制为30,000。

序列来源
可以从序列文件,序列通道和DNA数据库中检索序列片段数据。按“添加文件”按钮可从磁盘文件中检索序列,从文件夹中检索“文件夹”按钮(文件夹中的所有文件),从通道中检索“通道”按钮,或从默认数据库中检索“数据库”按钮。您可以通过选择它并按“删除”按钮从列表中删除序列,或者通过按“清除”按钮删除列表中的所有序列。
用于汇编的序列文件应采用带有ORIGIN关键字的DNAMAN格式(参见第IV.3节。如果您的任何序列文件没有ORIGIN格式,请检查加载整个文件,如果格式未知选项。跟踪文件(ABI和SCF)衍生自自动测序也在序列组装中被接受.DNAMAN还接受FASTA,GenBank,SAF和MSD格式的文件。
DNAMAN可以去除DNA片段的不确定区域。当ACGT%<选项时,您可以检查移除侧翼区域并设置百分比值。当此选项打开时,DNAMAN将计算所有输入序列的侧翼区域的组成。 ACGT%较低的区域将被删除。
您可以选择从所有源序列中删除矢量序列。在这种情况下,您必须将矢量序列加载到默认序列通道中。如果源序列有许多向量,建议将所有向量组合在一个序列文件中,并将其加载到默认序列通道中。当此选项打开时,DNAMAN将比较所有输入序列与矢量。将除去含有载体序列的侧翼区。矢量序列越短,比较越快。因此,您可以通过删除向量中不必要的序列来加速该过程。
使用排序项目时,建议为项目创建数据库。您可以在数据库中保存所有序列以及项目的向量。数据库方法使您的测序项目井井有条,便于序列组装。
序列组装方法
DNAMAN分两步组装序列。第一步是对齐以产生装配分数。第二步是使用分数矩阵组装片段。要生成对齐分数矩阵,您可以选择快速对齐或结束比较方法进行序列组装。在序列组装的大多数情况下,建议使用快速对齐方法。
结束比较方法逐步比较每个片段的结尾。没有插入序列结束。根据资格标准确定重叠。结束比较方法应仅用于少量碎片,而重叠短且重叠质量高。
使用快速比对方法,使用快速比对算法(Wilbur和Lipman,1983,Proc.Natl.Acad.Sci.USA,80:726-730)进行比对,所有序列均在正链和负链中。序列将根据资格标准进行组装。如有必要,将在重叠区域添加插入。最终装配有两种方法:快速和最佳。快速方法使用Wilbur&Lipman算法,可在短时间内完成装配。优化方法使用Smith&Waterman并提供更好的装配效果。大多数情况下建议使用最佳方法。组装序列有两种方法:从头和重新测序。重新排序将使用列表中最长的序列作为模板,并将所有其他序列组装到模板中。 de novo方法不使用任何模板,并将尝试根据分数矩阵对齐所有序列。如果模板可用,建议使用重新排序方法,因为它提供了最佳结果并使用较少的计算资源。
对齐参数是:
重叠中的最小核苷酸数。默认值为80个碱基。
重叠区域中的最小身份百分比。默认级别为90%。
快速对齐方法的参数。 k元组的缺省值,缺口开放罚分,缺口延伸罚分,窗口大小和对角线分别为4,6,0,4和6。这些设置对于一般序列组装是最佳的。
结束比较方法:最大重叠。该参数定义任何序列对的重叠区域的最大长度
装配分析
单击“组合”按钮开始搜索重叠序列。 DNAMAN首先对所有输入序列进行成对比较,并不断更新结果。指示在搜索期间找到的重叠数。序列组装的过程是线程化的,因此,您可以在序列组装期间执行其他任务。
处理序列组装的选项。
运行时最小化窗口。处理序列组装时,“组装”对话框将最小化。如果您想在序列组装期间执行其他任务,可以选中此选项。
重新排序序列。序列顺序将从原始输入顺序更改。每个重叠群中的所有序列将组合在一起。
DNAMAN将文件信息保存在汇编结果中。如果使用跟踪文件,则在序列组装编辑器中单击序列库时,它将显示跟踪文件中的位置。
您可以通过按“停止”按钮在装配过程中随时停止装配。
如果所有序列都不能组装成一个连续序列,它们可以分成不同的重叠群,这些重叠群也在组装过程中指示。在组装结果中将忽略隔离的序列。单击“显示结果”按钮以在序列组装编辑器中显示结果。
序列组装编辑器
序列组装编辑器有助于组装序列的可视化和编辑。序列组合编辑器中有三个窗口:名称列表窗口,序列窗口和图形窗口。
名单列表窗口
所有组装的序列名称都显示在此窗口中。相应的序列位于“序列”窗口中的名称旁边。您可以通过拖放名称来更改这些序列的显示顺序,以便向上或向下移动它们。您也可以通过双击更改任何序列的名称。
序列窗口
所有序列都列在此窗口中。共识序列位于窗口顶部。您不能直接编辑共有序列,但您可以通过编辑每个重叠序列来修改它。源序列的任何修饰都可能导致共有序列的改变。
汇编序列的编辑功能如下:
1)添加或去除碱
2)添加或删除间隙
3)选择一个序列的块并将其删除。
4)将所选序列更改为大写字母
5)将所选序列更改为小写字母
6)在项目中找到一个序列。
图形窗口
图形窗口包含序列组装图。图中有三个元素。

1)直线代表共有序列。共有序列的长度和位置在直线的两端表示。
您可以将光标放在共识序列的开头,在图形窗口中移动整个图表。当光标切换到时,按下鼠标左键,然后将图表拖放到所需的位置。您还可以更改直线的长度(一致序列)。将光标放在共识序列的末尾。当光标切换到东西向箭头形状时,按下鼠标左键,然后将结束拖放到任何位置
2)如果图中出现矩形,则表示共有序列中断。矩形用于划分不同的序列组。您可以通过向左或向右移动来使中断更大或更小。在这种情况下,“序列”窗口中的序列将根据您的调整移动。
3)每个源序列显示为箭头线。箭头表示序列的方向。通过将光标指向箭头线,您可以向上或向下移动它。所有输入序列的名称可以显示在面板的箭头或左侧。
序列组装编辑器的选项
单击“图形”窗口中的“选项”按钮以选择编辑器的选项。

显示双链序列。共有序列和所有输入序列以双链形式显示。
在图表中显示序列名称。 DNAMAN可以在图形面板的左侧或序列箭头上显示名称。
显示翻译。 DNAMAN可以在三个或六个阅读框中翻译共有序列。
序列导出
单击“图形”窗口中的“导出”按钮以在文本窗口中导出序列。您可以选择仅为短装配导出所有序列或共有序列。长组件仅将导出共有序列。
序列搜索
DNAMAN提供了许多搜索序列的方法。 您可以搜索一组DNA或蛋白质序列,并在图形窗口中显示结果。 您还可以搜索直接重复,镜像重复,反向重复或茎环结构。 DNAMAN还搜索给定DNA序列的氨基酸序列和可能的开放阅读框。
搜索核苷酸序列
DNAMAN从当前DNA序列的两条链中搜索核苷酸序列,并在图形窗口或文本窗口中显示搜索结果。
选择搜索| 用于打开“搜索”窗口的序列命令,您可以在其中搜索核苷酸序列和定义的共有序列。

查询格式
单击“查询”按钮将打开“输入序列”对话框。输入要搜索的核苷酸序列。有几种搜索格式(字母不区分大小写):
类型:AGGCGATG搜索:AGGCGATG
类型:AGGCNNNGATG搜索:AGGCNNNGATG(N = A,C,G或T)
类型:AGGC(N3)GATG搜索:AGGCNNNGATG
类型:AGGC(N3-10)GATG搜索:AGGC和GATG,它们之间有3-10个核苷酸。
类型:AGGC(X)GATG搜索:AGGC和GATG,它们之间有任意数量的核苷酸。
类型:AGGC [ACGT / TCGA] GATG搜索:AGGCACGTGATG或AGGCTCGAGATG
您也可以使用IUPAC代码搜索核苷酸。选择信息| Nucleotides命令显示IUPAC代码表。
例如:
类型:AGGWCGAT(W = A或T.)搜索:AGGACGAT或AGGTCGAT
例如,在“搜索”对话框中键入“GATT”,然后单击“确定”按钮以显示找到的序列列表。
DNAMAN允许您搜索多个序列并列出所有站点。再次单击“查询”按钮以打开“搜索”对话框。输入另一个序列,例如AATAAA,在对话框中单击“确定”按钮。 DNAMAN将在搜索结果列表和搜索图中添加找到的序列。找到的序列后面的数字表示查询序列的组。
单击“导出”按钮可在文本窗口中显示搜索结果。您可以使用打印命令打印文本。
搜索窗口中有两个面板:序列列表和图形显示。
序列表窗口
“序列列表”窗口包含四列。
位置:子序列的位置。该位置旁边有一个复选框。您可以选中并取消选中该框。如果选中,则后续位置将在图形演示中指示。
Strand:子序列所在的+或 - strand。
顺序:子序列及其侧翼区域。侧翼区通常在子序列的上游和下游5个碱基。
组:子序列所属的组和初始查询序列。您可以在目标序列上搜索许多查询。每个查询都会产生属于同一组的许多子序列。
您可以通过调整相应的标题大小来更改每列的大小。
图形演示
DNAMAN还以图形显示方式显示搜索结果。演示文稿中有两行;上面一行(Sequence Line)表示目标序列,下面一行(Zoom Line)表示上面一行的缩放。
图形演示是一般图形文档。您可以使用第V章(添加文本对象,复制图形......)中描述的工具来处理它。
所有找到的子序列的位置显示在序列线上。不同的子序列组用不同的颜色表示。如果在“序列列表”窗口中选中了子序列,则其位置将用箭头标记。
如何缩放:将光标放在两条长垂直线中的一条上,光标切换到向上箭头。通过移动箭头,您可以将焦点放在较小的区域上。在序列线上滑动向上箭头时,位置显示在屏幕的左上角。
如何显示序列:将光标放在序列线或缩放线上。移动时按住鼠标左键可选择一个区域。松开鼠标按钮,会出现一个对话框,显示所选序列。
如何移动图形:将光标放在序列线的左端。当光标切换到时,按下鼠标左键,然后将图表拖放到您想要的任何位置。您可以通过移动缩放线来更改两条线的相对位置。将光标放在缩放线的左端,按鼠标左键,然后将缩放线拖放到适当的位置。
如何调整图形大小:您可以更改序列线和缩放线的长度。将光标放在序列线或缩放线的右端。当光标切换到时,按下鼠标左键,然后将线拖放到适当的位置。
您可以通过单击Rem删除已检查的子序列。检查按钮,或使用Rem清除列表。全部按钮。
序列搜索选项
单击“选项”按钮将打开“序列搜索选项”对话框。您可以修改以下参数:
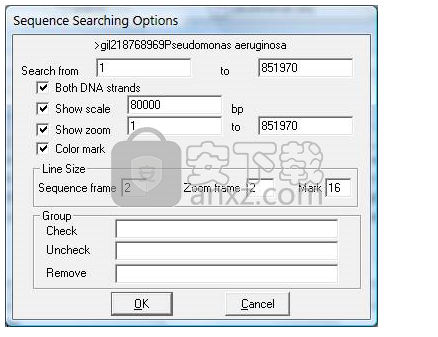
搜索区域。您可以搜索当前序列的全部或部分内容。定义搜索区域的开始和结束位置。请注意,缩放区域应位于定义的搜索区域内。
DNA都是两条链。您可以通过选中选项在当前序列的两条链上搜索核苷酸序列。如果只想搜索感测序列,请取消选中此选项。该选项不适用于蛋白质序列。
显示比例。您可以通过选中显示比例选项在搜索图上显示比例。可以修改比例尺寸。
显示缩放。您可以通过选中/取消选中“显示缩放”选项,在搜索图上显示或隐藏缩放线。变焦位置可以更改为所需的值。
颜色标记。通过选中“颜色标记”选项,在图表中以不同颜色标记搜索结果。
线条大小。您可以更改“序列线”和“缩放线”的线宽。可以在“标记高度”框中修改每个标记的大小。
组。您可以在子序列列表中选中/取消选中一个或多个组。您还可以从列表中删除一个或多个组。在相应的框中输入组的编号。如果输入多个数字,则数字应以空格分隔。
寻找共识序列
您可以在图形搜索窗口中搜索共有序列,例如启动子和调控因子结合位点。
单击“共识”按钮将打开“共识序列”对话框。您可以通过单击序列名称来选择个体共识。相关信息将显示在“序列信息”框中。您还可以通过单击“全部搜索”按钮搜索共识序列的完整列表。
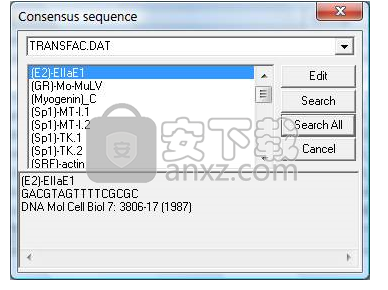
共有序列信息存储在数据文件中。您可以单击“编辑”按钮对其进行编辑。编辑数据文件的格式很简单。每个共有序列的信息由“//”分隔,由三行组成,即名称,共有序列和相关信息。编辑完成后,选择File |保存命令以保存更改。
IUPAC代码可用于共识序列。
搜索一组序列
您可以搜索一组序列,例如共识站点列表。结果以文本格式显示。
选择搜索|核苷酸序列集或蛋白质序列集命令。将出现一个对话框,您可以在其中选择包含搜索列表的文件。
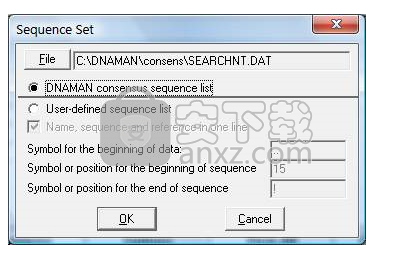
单击“文件”按钮以浏览并选择列表文件。列表文件可以是DNAMAN一致格式或用户定义格式。如果选择了DNAMAN共有序列表,则可以忽略其后的参数。 DNAMAN共识文件的样本可以在DNAMAN程序的CONSENS文件夹中找到。如果选择了用户定义的序列列表,则应输入有关列表文件格式的信息。 DNAMAN根据提供的信息提取列出的序列。
名称,顺序和参考在一行。如果每行定义文件中的序列,请选中此选项。
数据开头的符号。列表文件可能包含序列列表之前的一些必要信息。符号,例如“ORIGIN”,用于分隔信息和序列列表。
序列开头的符号或位置。如果名称,序列和参考在一行中输入序列的起始位置。否则,请输入关键字。
序列结束的符号或位置。如果名称,序列和参考在一行中输入序列的结束位置。否则,输入关键字或什么也不输入。
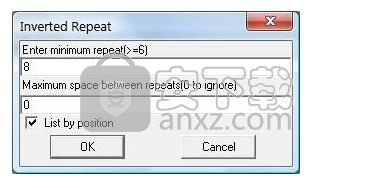
搜索反转重复(词干结构)
DNAMAN在当前序列中搜索可能形成茎环/发夹结构的互补序列。选择搜索|反转重复菜单。有两个与搜索相关的参数:发夹结构的茎中成对核苷酸的最小长度(碱基)和互补序列之间未配对核苷酸的最大长度(碱基)。
寻找氨基酸序列
DNAMAN从当前序列的六个阅读框中搜索氨基酸序列(一个字母代码)及其变体。
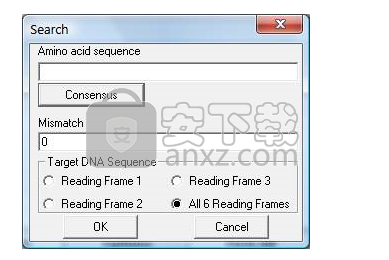
错配可以是氨基酸序列中的任何位置。
例如:
键入VXPSC并将不匹配参数设置为0以搜索VXPSC,其中“X”对应于位置2处的未定义氨基酸。
键入VSPSC并将错配参数设置为1,在这种情况下,错配氨基酸可以在任何位置。
蛋白质共有序列
单击“搜索”对话框中的“共识”按钮以打开共识序列框。选择一个共识序列,它将被加载到“搜索”对话框中。您可以编辑共识列表。氨基酸序列的编辑方法与DNA共有序列的编辑方法相同。有关文件格式,请参阅DNAMAN程序的CONSENS文件夹中的SEARCHNT.DAT文件。
寻找开放阅读框架
您可以从当前序列的六个阅读框中搜索开放阅读框(ORF)。默认情况下,DNAMAN使用通用遗传密码表进行搜索。在某些情况下,您可能希望使用CTG而不是ATG或两者作为起始密码子。 DNAMAN允许您通过选择Protein |来进行这些更改遗传密码表命令然后选择适当的遗传密码表(参见XI.1 XI.1部分)。 DMANAN将根据所选遗传密码表中的起始密码子和终止密码子搜索ORF。
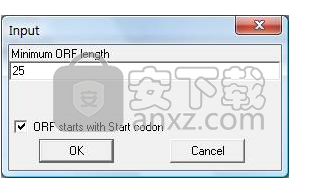
DNAMAN仅显示每个阅读框中的两个最大的ORF。
SportTracks(跑步记录分析) v3.1.4938 中文 健康医药5.00 MB
详情健康自测 1.0 健康医药0.07 MB
详情《日常生活禁忌全书》扫描版 健康医药20.00 MB
详情GraphPad Prism 8(医学绘图软件) 健康医药29.4 MBv8.0.0
详情准妈妈乳母营养师 健康医药3.00 MB
详情7分钟锻炼 v1.347 去广告 健康医药18.00 MB
详情medcalc注册机 健康医药0.54 MB附使用教程
详情华佗中医大师 v4.9 健康医药14.00 MB
详情medcalc 19(医学统计软件) 健康医药58.1 MBv19.5.6 中文
详情Cookbook+Calendar(饮食计划软件) 健康医药16.7 MBv3.9
详情护眼小精灵 1.4 健康医药0.25 MB
详情生活宝典 疾病医疗与健康 健康医药12.00 MB
详情医学三基考试宝典(医师) 3.0 健康医药5.00 MB
详情dnaman(序列分析软件) 健康医药14.7 MBv9.0
详情MasterCook 2020(食谱管理软件) 健康医药285 MBv20.0.1.1 附安装教程
详情牙医管家(口腔管理软件) 健康医药182.22 MBv5.2.500.1 官方版
详情《默克家庭诊疗手册》 健康医药0 MB
详情《玉房秘诀》 健康医药0.47 MB
详情《男性增强术》 健康医药0 MB
详情《家庭医生2006》注册版 健康医药75.00 MB
详情《玉房秘诀》 健康医药0.47 MB
详情《中医特效秘方绝技》外科卷 健康医药0.41 MB
详情你想知道而不敢问的性知识 健康医药0.54 MB
详情人体穴位图合集 彩图高清版 健康医药55.00 MB
详情《家庭医生2006》注册版 健康医药75.00 MB
详情medcalc 19(医学统计软件) 健康医药58.1 MBv19.5.6 中文
详情《民间实用土方》 健康医药0.17 MB
详情《本草纲目》 图解版 健康医药18.00 MB
详情家庭用药指南.3.0.1.2 特别版 健康医药4.00 MB
详情《夫妻幸福百事通》PDF 健康医药0.54 MB
详情中国美食菜谱 1.05 绿色版 健康医药5.00 MB
详情《精神病学教科书(第五版)》 PDF 健康医药178.00 MB
详情《男性增强术》 健康医药0 MB
详情《力量与肌肉训练图谱》PDF扫描版 健康医药47.00 MB
详情《茶包小偏方喝出大健康》 PDF版 健康医药332.00 MB
详情社保费代收客户端 健康医药18.3 MBv1.0.003 官方版
详情《超级体格强壮术》 健康医药3.08 MB
详情《人体穴位与推拿》 健康医药3.35 MB
详情《中国的野菜》(319种中国野菜图鉴)扫描版 健康医药67.00 MB
详情《穴位按摩速查全真图解》扫描版 健康医药34.00 MB
详情《95%的人都不知道的养命方》扫描版 健康医药39.00 MB
详情《默克家庭诊疗手册》 健康医药0 MB
详情《男性增强术》 健康医药0 MB
详情护眼小精灵 1.4 健康医药0.25 MB
详情针灸中医临床管理 ShenProfessional 3.1 中文绿色专业版 健康医药57.00 MB
详情《催眠入门手册》 健康医药0 MB
详情眼睛卫士(EyeGuard) 3.01 Final 健康医药1.00 MB
详情人体生物节律速查器 优生优育、择日 绿色版 健康医药4.35 MB
详情《民间实用土方》 健康医药0.17 MB
详情怀孕完全知识手册 健康医药0.09 MB
详情